(来源:DrugAI)
DRUGONE
在当前的大规模单细胞组学研究中,数百个样本常被用于解析复杂的疾病或生物过程。然而,传统分析方法通常通过对细胞信息进行平均处理来简化样本层级的差异,从而掩盖了关键的细胞群体变化。为此,研究人员开发了一种新型深度生成模型——多分辨率变分推断(Multi-resolution Variational Inference, MrVI),用于在单细胞层级上解析样本间的异质性。
MrVI同时处理两个关键任务:样本分群(exploratory analysis)与差异比较(comparative analysis),无需预先定义细胞类型。通过在单细胞层面建模样本协变量,MrVI揭示了仅在特定细胞群体中体现的疾病相关变化。例如,在新冠患者和炎症性肠病(IBD)人群中,MrVI识别出了此前被忽视的细胞亚群及其分子特征。此外,MrVI还能在大规模药物扰动实验中自动识别具有相似生化效应的小分子群。该模型已作为开源工具在 scvi-tools.org 上发布。

在过去十年中,大规模功能基因组学推动了研究人员对临床、遗传和环境因素如何在细胞层面上表现的理解。如今,随着单细胞测序技术的成熟,这些研究正进入更高分辨率阶段。单细胞组学能够揭示样本内部和样本之间的复杂差异,为疾病亚型识别和治疗策略提供新的契机。
然而,传统的分析策略主要面向小规模样本研究,强调细胞间差异;在面对数百样本级别的单细胞数据时,这种方法变得不再适用。样本层级分析主要包括两类任务:
探索性分析(exploratory analysis):根据细胞和分子特征将样本自动分组,以发现未知的亚群;
比较性分析(comparative analysis):在已知的样本分组间识别差异性表达(DE)基因或差异性丰度(DA)细胞群。
目前的主流方法依赖于预定义的细胞聚类或注释,这在数据复杂、异质性强的多样本研究中表现受限。此外,不同细胞群之间的变化常被平均化,导致仅在特定细胞亚群中存在的疾病信号被忽略。
为克服这些问题,研究人员提出了MrVI框架,它在无注释条件下实现跨样本的分层建模。该模型能在保留细胞分辨率的前提下完成样本分群与差异分析,适用于跨个体、跨组织或跨实验条件的研究。
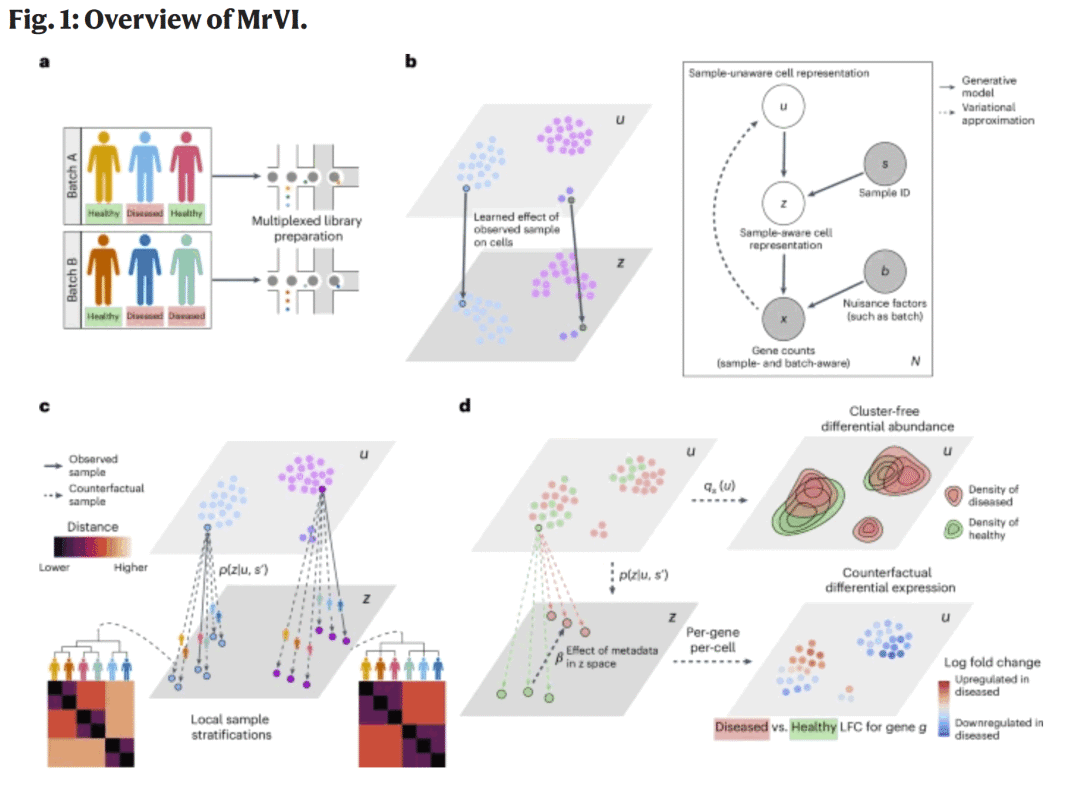 图1. MrVI框架概述
图1. MrVI框架概述方法
MrVI是一个分层贝叶斯生成模型,用于多样本单细胞RNA测序数据的综合、探索与比较分析。模型区分两类协变量:
目标协变量(target covariate):反映感兴趣的样本特征,如个体ID、处理条件或疾病状态;
噪声协变量(nuisance covariate):反映技术或实验来源,如测序批次或建库方法。
每个细胞被映射为两个低维潜变量:
u 表示样本无关的细胞状态,捕捉共享的细胞类型信息;
z 则结合目标协变量,反映样本引起的变化。
MrVI使用高斯混合分布作为先验,通过深度神经网络完成编码与解码。模型训练过程基于变分推断框架,最大化证据下界。
在推理阶段,MrVI支持两类分析:
探索性分析:通过计算“样本距离矩阵”,评估细胞在不同样本假设下的潜在表示,从而自动识别样本分群。
比较性分析:通过反事实推断估计在不同样本条件下的基因表达分布,实现无注释的DE和DA检测。
这种设计允许MrVI同时捕获细胞状态变化与样本层级效应。
结果
半合成数据验证
研究人员首先在模拟的外周血单核细胞(PBMC)数据上验证MrVI的准确性。结果显示,MrVI能够在不同细胞亚群中同时识别DE与DA效应,并正确重建样本间的层级关系。与传统基于聚类的分析相比,MrVI在无注释条件下获得了更高的识别精度与召回率。
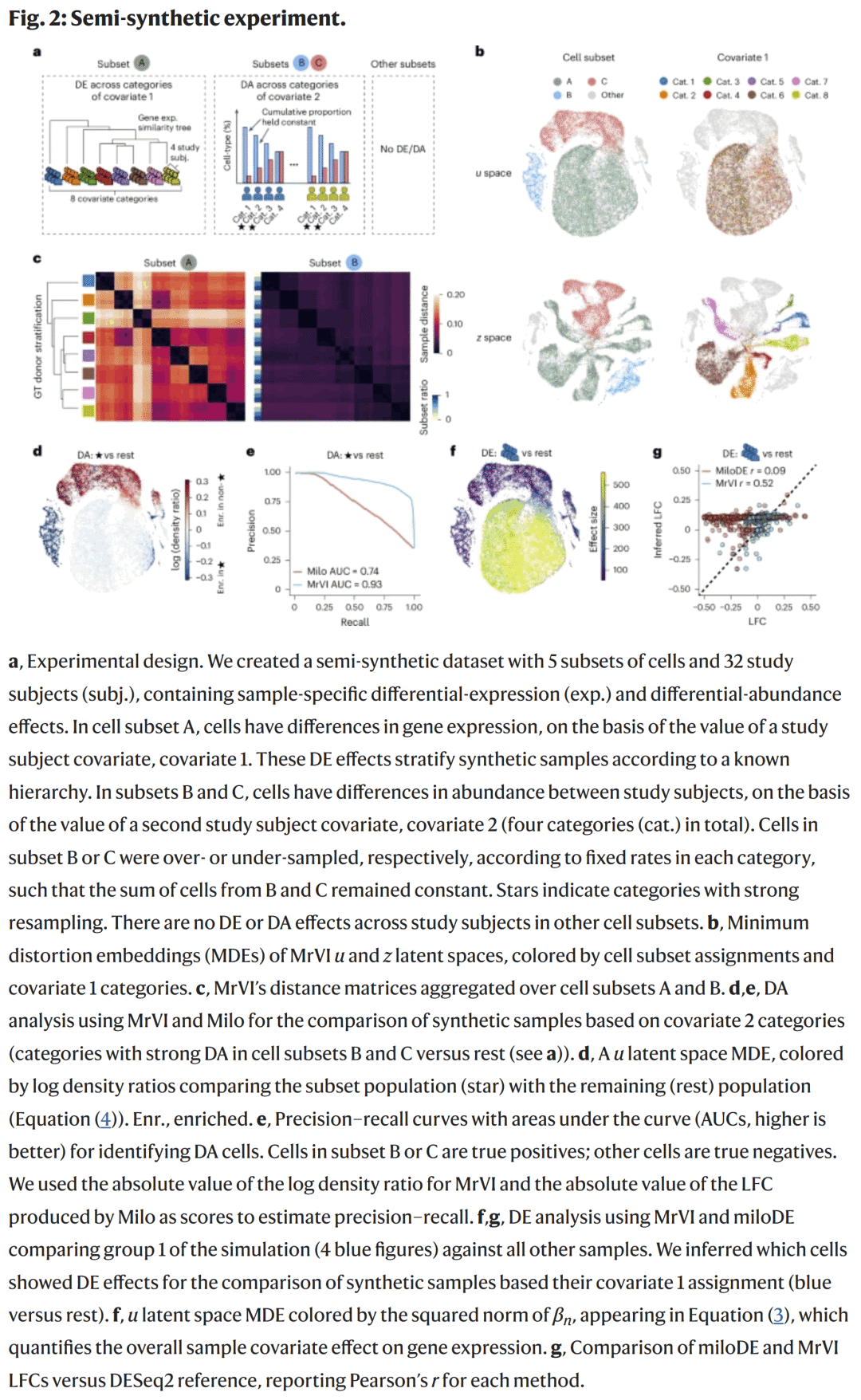 图2. 半合成数据实验结果
图2. 半合成数据实验结果新冠患者免疫异质性分析
研究人员将MrVI应用于来自419,000个PBMC的COVID-19队列数据。模型在细胞类型分辨率上实现了样本整合,并揭示了单核细胞中特异性免疫反应。通过“反事实距离矩阵”,MrVI不仅区分出健康人与感染者,还进一步将感染者按症状持续时间分为两个亚群。后续分析显示,这种分群与单核细胞活化状态及感染时间高度相关。
差异分析发现,疾病早期患者的CD16⁺单核细胞与树突细胞显著减少,而感染后期个体的炎症基因(如TNF、NFKBIZ)上调,提示免疫状态的时间依赖性变化。
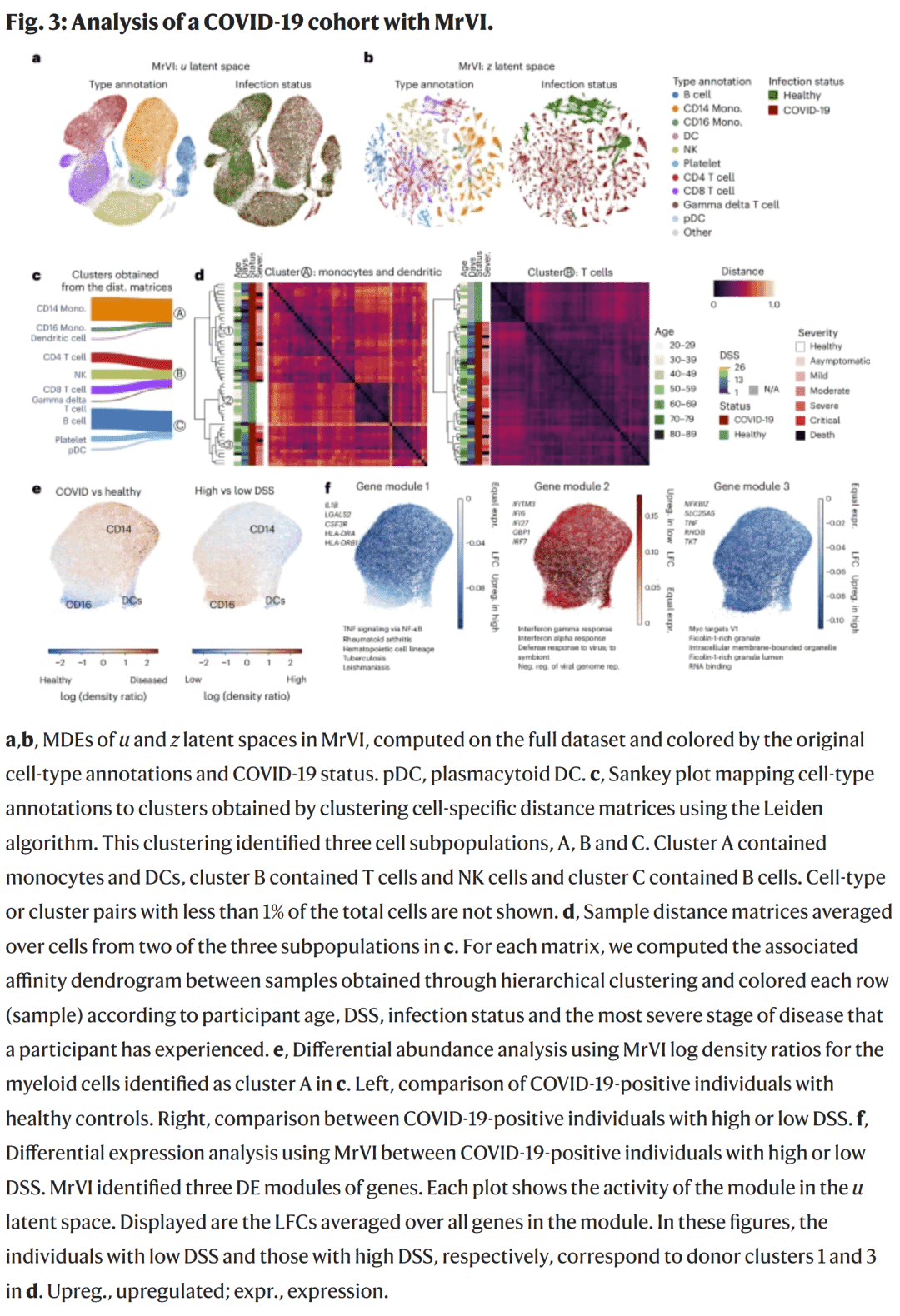 图3. COVID-19队列中免疫细胞的分群与差异分析
图3. COVID-19队列中免疫细胞的分群与差异分析化学扰动实验中的药物效应聚类
在sci-Plex单细胞药物筛选数据中,研究人员使用MrVI分析188种小分子化合物的转录组效应。模型在整合重复实验的同时,自动识别出具有相似机制的药物群。例如,HDAC抑制剂形成了独立簇,与代谢下调途径显著相关,而MEK抑制剂trametinib被正确聚类至RAS信号通路模块。
与基准方法相比,MrVI在药物相似性度量上表现最佳,成功捕获剂量依赖关系及新的机制相关性。
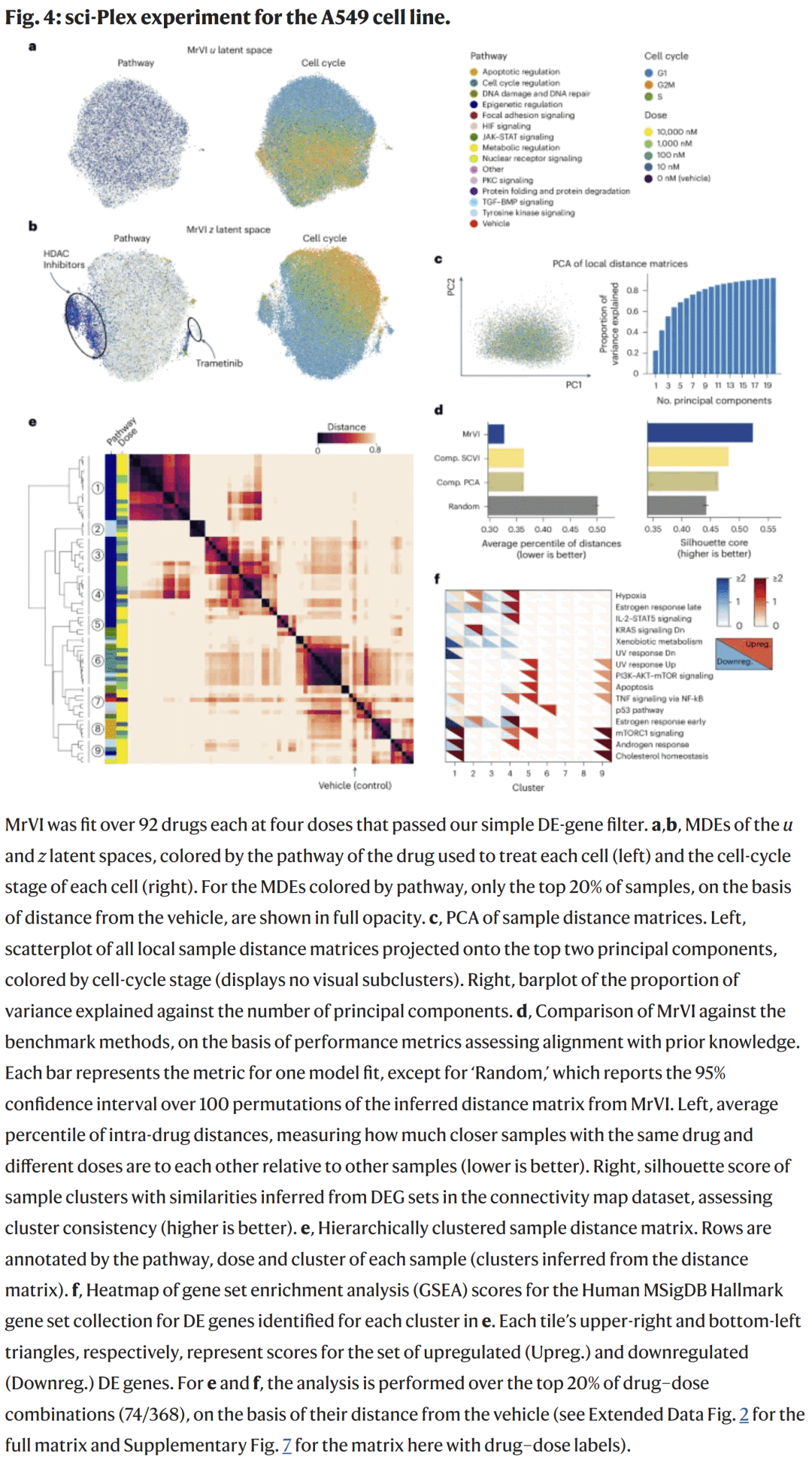 图4. 药物扰动实验中的MrVI分析
图4. 药物扰动实验中的MrVI分析炎症性肠病(Crohn’s disease)队列分析
在分析46例Crohn’s患者和25名对照者的单细胞RNA测序数据时,MrVI揭示了与肠道狭窄(B2型病变)相关的间质细胞变化。模型发现:
B2型患者中,血管内皮细胞数量减少,而成纤维细胞与周细胞群显著增加;
在HIGD1B⁺STEAP4⁺周细胞亚群中,CDH11、COL1A1、TGFBI等纤维化标志物显著上调;
某些内皮细胞出现了向周细胞样状态转变,提示炎症组织中存在血管–间质过渡现象。
这些发现为炎症性肠病的分型与潜在治疗靶点提供了新线索。
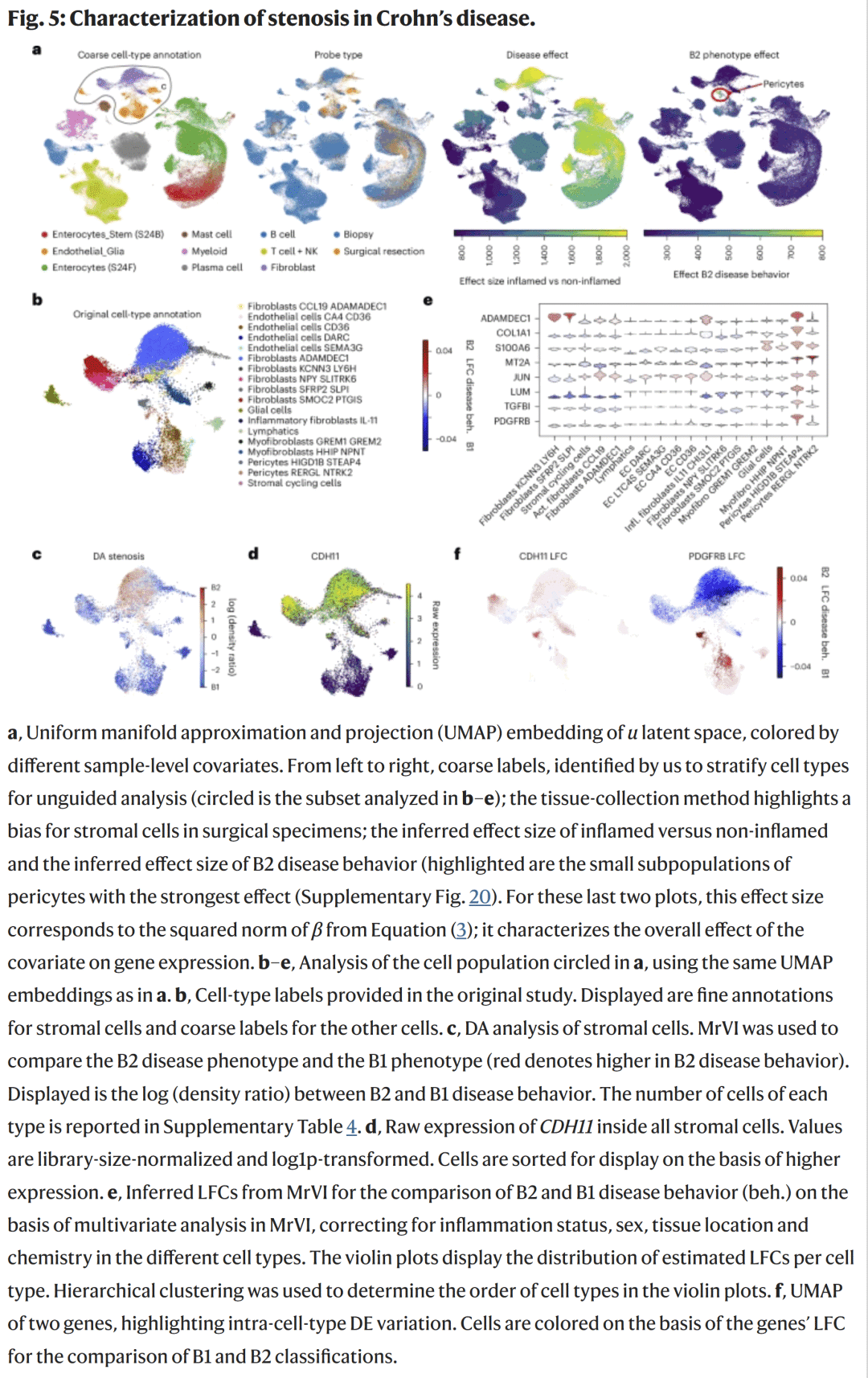 图5. Crohn’s病中与狭窄相关的细胞变化
图5. Crohn’s病中与狭窄相关的细胞变化讨论
MrVI提供了一个统一的深度概率框架,可在单细胞层级解析样本间异质性。该方法无需细胞类型注释即可完成样本整合、分群与差异分析。相比传统基于聚类或假设分组的方法,MrVI具备以下优势:
能从下而上地推断样本分层结构,揭示潜在疾病亚型;
能在细胞层级上量化差异表达与丰度变化;
具备强大的扩展性,可应用于多模态数据(如ATAC、蛋白组等)。
研究人员指出,未来的改进方向包括:
引入跨模态建模机制,以捕获染色质状态与转录变化间的关联;
优化模型结构以支持上百万细胞级别的多样本整合;
将MrVI扩展至跨物种或多条件的生物学比较研究。
总体而言,MrVI为解析样本层级异质性与疾病多样性提供了一种强大且通用的深度生成建模框架。
整理 | DrugOne团队
参考资料 ]article_adlist-->Boyeau, P., Hong, J., Gayoso, A. et al. Deep generative modeling of sample-level heterogeneity in single-cell genomics. Nat Methods (2025).https://doi.org/10.1038/s41592-025-02808-x

内容为【DrugOne】公众号原创|转载请注明来源
]article_adlist--> 海量资讯、精准解读,尽在新浪财经APP
海量资讯、精准解读,尽在新浪财经APP
证券配资公司,配资最良心10大平台,如何选择证券公司提示:文章来自网络,不代表本站观点。
- 上一篇:免息炒股配资安排了连续的四个场面
- 下一篇:炒股票软件排名淘宝天猫押注投入AI产研